COVID-19 Vaccines: When Will the Pandemic End?

As Pfizer, Moderna, and other pharma companies prepare to seek emergency authorization for their SARS-CoV-2 vaccines, the FDA has laid out a roadmap designed to ensure appropriate scientific rigor and help engender public trust. That plan was the subject of a special meeting of the Vaccines and Related Biological Products Advisory Committee (VRBPAC) on October 22, 2020, where experts discussed 2 critical FDA guidance documents that provide a blueprint for development and approval of SARS-CoV-2 vaccines. That blueprint is at the center of a massive government effort to quickly and safely speed vaccines to the American public and bring the pandemic to an end.
Members of the VRBPAC, with expertise in infectious disease, epidemiology, and vaccine development, focused on issues around the FDA standards for safety and effectiveness that will support Emergency Use Authorization (EUA) of vaccine candidates. They discussed the need to continue the phase 3, randomized, placebo-controlled trials to completion after an EUA is granted. They considered how the vaccines will be rolled out to the American public, and they raised concerns about whether the public will embrace the vaccines and roll up their sleeves.
The committee acknowledged that this is a high-stakes game, and if we lose public trust, it will fail.
Representatives from government agencies responsible for managing Operation Warp Speed (OWS), including the FDA, the Centers for Disease Control and Prevention (CDC), the National Institutes of Health (NIH), and Health and Human Services’ Biomedical Advanced Research and Development Authority (BARDA), provided an overview of the operation. Those presentations highlighted the unprecedented level of engagement and collaboration that is involved in OWS. Never has there been such a massive and coordinated effort on the part of the government to launch such an ambitious vaccine program in such a short period of time.
Robert Johnson from BARDA outlined the 6 lead vaccine candidates that are likely to be approved for emergency use in the coming months. Those include: 2 mRNA vaccines (a novel technology), being developed by Moderna and by BioNTech in collaboration with Pfizer; 2 adenovirus vaccines, being developed by Janssen and by Oxford University in collaboration with AstraZeneca; and 2 protein subunit vaccines, being developed by Novavax and by a partnership between GlaxoSmithKline and Sanofi.
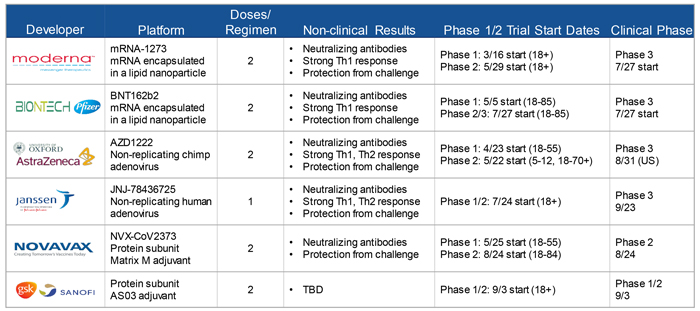
Source: Robert Johnson, PhD, Director, Division of Influenza and Emerging Infectious Diseases BARDA/ASPR/HHS, presented at VRBPAC meeting, October 22, 2020. https://www.fda.gov/advisory-committees/advisory-committee-calendar/vaccines-and-related-biological-products-advisory-committee-october-22-2020-meeting-announcement
While the VRBPAC agreed that the FDA guidance is appropriate and the OWS plan is sound, many advisors and members of the public, who spoke at the meeting, raised questions and concerns about the rigor of the interim safety and effectiveness data that will support an EUA. The public sentiment is that the vaccines are being developed too quickly, leading some to question whether full approval rather than EUA might be the best option to engender public trust. Another important topic of discussion was whether the clinical trials are enrolling enough racial and ethnic minorities, older individuals, and those at highest risk of developing severe complications from COVID-19. Certain groups, including children and pregnant women, are not currently being enrolled in the clinical trials.
So, where do we go from here? Pfizer and Moderna have recently announced that they have interim data showing that their vaccines are more than 90% effective with no serious safety concerns. The next step will be for those companies, and others to follow, to apply for an EUA. The FDA will then convene a meeting of the VRBPAC to review the data and make a recommendation for, or against, an EUA.
Both Pfizer and Moderna hope to meet with the VRBPAC before the end of December and that FDA will then quickly grant an EUA. In the meantime, both companies are continuing their ongoing phase 3 trials and will continue gathering more data to support full approval. They are already producing millions of doses of their vaccine and could make some available by years’ end, if approved. If all goes well, limited distribution to healthcare workers and other high-risk individuals could begin in mid to late December.
The question of when the majority of Americans will have access to a vaccine is still hotly debated. Assuming that 3 or 4 of the top 6 candidates prove to be safe and effective by the first quarter of 2021, experts have estimated that the majority of Americans could have access by midyear. Whether vaccines can bring an end to this public health emergency depends largely on the public health strategy and whether the public is willing and able to get vaccinated. Everyone involved is doing their best to adhere to rigorous scientific standards and ensure that the vaccines are both safe and effective. Will we trust the science and roll up our sleeves?
About ProEd Regulatory: Since 1997, ProEd Regulatory has helped sponsors prepare for more than 150 FDA Advisory Committee meetings.

Jeff Riegel, PhD, combines his scientific expertise with more than 25 years of global healthcare agency experience in guiding medical and regulatory communication strategies for biopharma companies. Jeff helps clients prepare for FDA Advisory Committee meetings and other health authority interactions. Connect with Jeff on LinkedIn.