Can You Predict Whether You Will Face an FDA Advisory Committee?
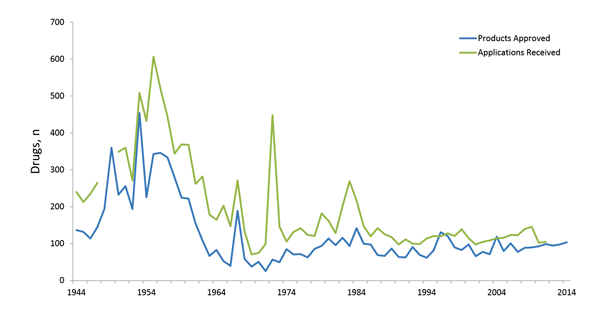
 The US Food and Drug Administration (FDA) review of drugs, although guided by broad standards, remains individualized. Each drug or drug class faces unique challenges during the FDA review phase – thus no two reviews are exactly the same. Each review division within the FDA has its own approach, and evaluations often differ among the centers. To help understand the review process, the team at ProEd Regulatory sought to learn from the past to inform our future. We reviewed applications submitted to the Center for Drug Evaluation and Research (CDER) data over the last 70 years to see what patterns might emerge. Although the largest proportion of products (drugs and biologics) fails between discovery and Phase 3, an evaluation of applications submitted to the FDA over the last 70 years shows that the vast majority of products that reach the submission stage are ultimately approved (Figure 1).1
The US Food and Drug Administration (FDA) review of drugs, although guided by broad standards, remains individualized. Each drug or drug class faces unique challenges during the FDA review phase – thus no two reviews are exactly the same. Each review division within the FDA has its own approach, and evaluations often differ among the centers. To help understand the review process, the team at ProEd Regulatory sought to learn from the past to inform our future. We reviewed applications submitted to the Center for Drug Evaluation and Research (CDER) data over the last 70 years to see what patterns might emerge. Although the largest proportion of products (drugs and biologics) fails between discovery and Phase 3, an evaluation of applications submitted to the FDA over the last 70 years shows that the vast majority of products that reach the submission stage are ultimately approved (Figure 1).1
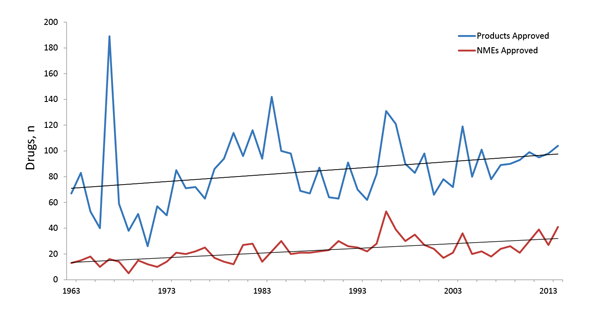
 It wasn’t until the Kefauver-Harris Drug Amendments of 1962 that the FDA required sponsors to demonstrate both efficacy and safety of their drugs prior to market access. In the wake of this significant legislation’s impact on drug development, we still see a consistent flow of approved applications and a corresponding subset of new molecular entities (NMEs) (Figure 2).1
It wasn’t until the Kefauver-Harris Drug Amendments of 1962 that the FDA required sponsors to demonstrate both efficacy and safety of their drugs prior to market access. In the wake of this significant legislation’s impact on drug development, we still see a consistent flow of approved applications and a corresponding subset of new molecular entities (NMEs) (Figure 2).1
The FDA approves each product based on its unique benefit:risk profile, which comprises the efficacy data, safety data, and the current need of the patients. The FDA began seeking external advice as early as 1964, and the Federal Advisory Committee Act of 1972 established a more formal process by which Federal agencies, such as the FDA, could seek outside expertise. In 1997, just before ProEd Regulatory was formed, President Clinton signed the Food and Drug Administration Modernization Act (FDAMA), which included instructions on how to conduct advisory committee meetings. Initially the FDA convened meetings frequently (~60 meetings annually).1,2 However, the number of CDER products reviewed by an FDA advisory committee has since declined and over the last 10 years has plateaued at approximately 40 per year (Figure 3).
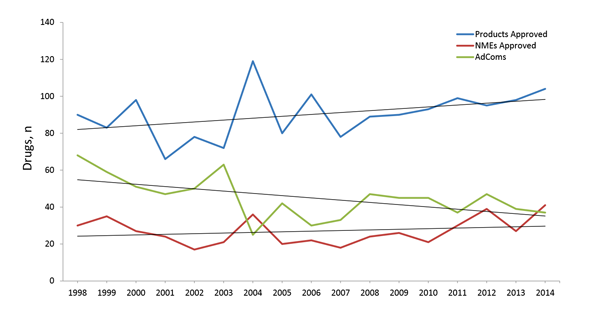
A review of the outcomes from recently convened advisory committee meetings indicates that the FDA does indeed rely on the input provided by these external panels. The concordance between the advisory committee vote and the final FDA decision is >85%.3 It is a common misperception that NMEs will automatically be reviewed by an advisory committee. This is because the Federal Food, Drug, and Cosmetic Act (FDCA) (21 U.S.C. 321; FDCA §§ 505A(i)(2)(A)) and guidance from the FDA in 2008 indicate that the FDA should carefully consider bringing all NMEs to an advisory committee for review. But in practice, between 2001 and 2010, only 37% of NMEs approved by the FDA were reviewed by an advisory committee.3 Similarly, in 2014, the FDA-approved 41 NMEs; however, only 14 or 34% of those drugs were reviewed by an advisory committee.1,2 If we take a closer look at those 14 NMEs approved in 2014, there is no clear cut category of drugs that were reviewed by an advisory committee.

Over the last 10 years, approximately one third of NMEs were reviewed by an advisory committee meeting, suggesting that all NMEs are not required to be reviewed by an advisory committee. And when you look over the same 10-year period, a similar percentage of all approved NDAs were reviewed by an advisory committee. So why did specific drugs face committees? The primary reasons are 1) matter of significant public interest, 2) controversial topic, or 3) need for special expertise.4 These typically translate to NMEs, understanding how a clinical trial with a novel design should be interpreted, evaluation of safety concerns, and assessment of overall benefit:risk profile. Taken together, these data confirm that there are no guarantees in drug development. Most, but not all, drugs that reach NDA status are approved, and there are no direct correlations between advisory committees and NMEs or special designations, such as orphan diseases. Therefore building your development plan on a solid foundation of science, with an understanding of the changing needs of patients, physicians, and healthcare providers is a must. So too, is the need to stay abreast of the changes in regulations and guidance that will undoubtedly impact your development program. ProEd Regulatory has been in front of the FDA and other health authorities for over 15 years, seeing the legislation and associated process changes as they evolve. Armed with this insight, ProEd can help you plan your path forward to regulatory and commercial success: www.proedregulatory.com.
References:
1. http://www.fda.gov/AboutFDA/WhatWeDo/History/ProductRegulation/SummaryofNDAApprovalsReceipts1938tothepresent/default.htm#Notes
2. http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/DrugandBiologicApprovalReports/NDAandBLAApprovalReports/ucm373413.htm
3. Ma P, et al. FDA Advisory Committee Outcomes. McKinsey & Company. 2013.
4. http://www.fda.gov/ucm/groups/fdagov-public/@fdagov-afda-gen/documents/document/ucm125651.pdf