Extending Asset Reach and Protecting Your IP Through the 505(b)(2) Pathway

Developing a novel pharmaceutical product from discovery to market launch can take up to 10 years and cost as much as $1 billion dollars1. The traditional 505(b)(1) approach to drug development involves a linear progression starting with nonclinical pharmacology, toxicology, and other PK studies, and typically culminates with large randomized phase 3 trials. This stepwise progression is time consuming and expensive, but essential to demonstrate the safety and efficacy of new molecules and gain FDA approval. Naturally, the cost associated with market entry discourages some smaller companies and restricts their focus to regulatory strategies with a lower financial barrier. The 505(b)(2) regulatory approval pathway presents a tangible opportunity for pharmaceutical companies to gain market entry and market share for a capital investment of $10-100 MM and 2-6 years of time. To put this into perspective, in 2019, approximately 65 505(b)(2) applications were filed in the United States compared with 48 505(b)(1) applications. In two parts, I will explore the utility of the 505(b)(2) pathway and offer insight into how both large and small pharma companies can capitalize on the innovative nature of this strategy.
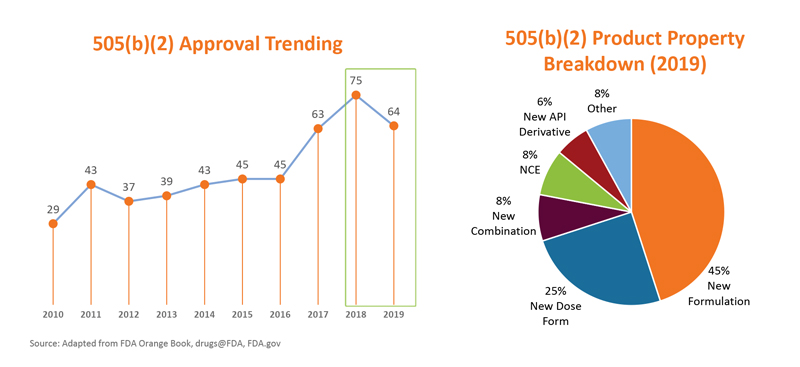
Part 1: Simple Explanation of the 505(b)(2) regulatory filing pathway
The 505(b)(2) pathway is a hybrid approach between the new chemical entity (NCE) and generic paths. The rigor and quality standards of the standard pathway still apply, but the 505(b)(2) pathway allows for utilization of pre‑existing data, such as safety data from a previous FDA filing or data from published literature. This encourages innovation by considering what an asset can do rather than what it was designed to do. The 505(b)(2) pathway can be used for changes in active pharmaceutical ingredient (API) form, new dosing formulations, new dose strengths, new indication(s), new fixed-dose combinations, and new drug/device combinations to satisfy an unmet medical need. This approach utilizes established safety data, requires shortened development time, and can drive innovation, solving problems through practicality. These innovations provide a lower financial barrier to market entry and provide real value to patients.
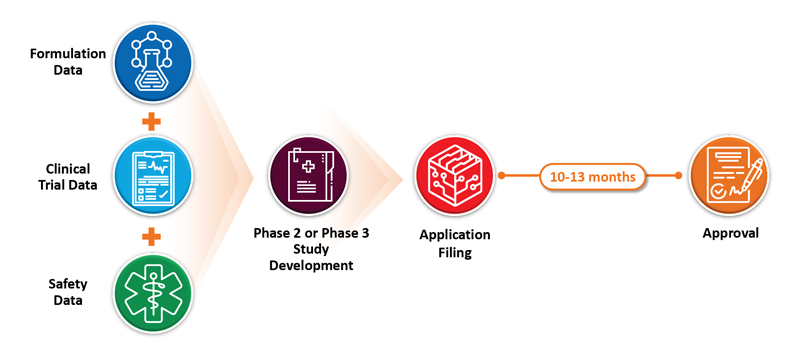
A key advantage of the 505(b)(2) strategy is that clinical trials are typically abbreviated and can run concurrently because of the available safety data on the approved drug. That means that the safety of the drug does not need to be re‑established, saving time and money. Depending on the drug, formulation and product configuration, the types of studies that need to be performed can vary. Let’s look at a couple of examples to see how this works. Narcan® for opioid overdose is Naloxone HCl in a nasal spray to replace injection and auto-injector configurations. Injection and an auto-injector limited the use of the medication to health care professionals (HCPs) and emergency personnel. Conversion to a nasal spray allowed the rescue therapy to be converted to an over the counter (OTC) product because of the ease of use, eliminating the need for trained professional administration. In that filing, the FDA only required a bioavailability (BA) study in healthy volunteers to demonstrate that the nasal spray was able to meet deliver relevant therapeutic levels with each use.

Another example is Zuplenz® Oral. This innovation converted Ondansetron for emesis given as an orally disintegrating tablet (ODT), to a patient-friendly sublingual film configuration. In this example, a dosing form change prompted bioequivalence (BE) studies to show that the sublingual film provided the same dissolution and performance capabilities of the ODT.

Up to this point, we focused on a high-level introduction to the 505(b)(2) pathway, contrasting it briefly against the traditional pathway. It can be inferred that small and mid-size pharmaceutical companies, startup organizations, investors, physician scientists and academic scientists can benefit from this strategy. The obvious advantage is speed to market and lower development costs facilitated by availability of existing data. In our next installment, we will focus on how to identify development opportunities and discuss the subtleties of critical early development activities, which make the 505(b)(2) pathway a unique approach to pharmaceutical development.
 Jeffrey Mocny, PhD, is a product strategy and development leader who leverages his experience in quality management, strategic planning, IND and NDA guidance, and risk mitigation to guide regulatory projects from conceptualization to completion in the analytical, medical device, biotechnology, nutraceutical, and pharmaceutical sectors. Jeff provides a consultative approach to sponsor relations, devises strategic roadmaps that follow best practices, and spearheads collaborative problem resolution. Connect with Jeff on LinkedIn.
Jeffrey Mocny, PhD, is a product strategy and development leader who leverages his experience in quality management, strategic planning, IND and NDA guidance, and risk mitigation to guide regulatory projects from conceptualization to completion in the analytical, medical device, biotechnology, nutraceutical, and pharmaceutical sectors. Jeff provides a consultative approach to sponsor relations, devises strategic roadmaps that follow best practices, and spearheads collaborative problem resolution. Connect with Jeff on LinkedIn.
Reference
- Wouters, Olivier J.; McKee, Martin; Luyten, Jeroen (2020-03-03). “Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009-2018”. JAMA. 323 (9): 844–853. doi:10.1001/jama.2020.1166. ISSN 0098-7484.