FDA Sets High Bar for Real-World Evidence in Rare Diseases
Real-world data (RWD) can be used to create historical control groups for clinical trials in rare diseases where a randomized controlled trial (RCT) is not feasible. But what happens when the US Food and Drug Administration (FDA) doesn’t accept it?
Since passage of the 21st Century Cures Act in 2016, FDA has promoted the use of real-world evidence (RWE) to increase the efficiency of clinical research. However, according to
FDA’s 2018 RWE framework, the use of RWE is primarily restricted to evaluating safety
(eg, monitoring postmarketing safety). It can only be used in limited circumstances to inform decisions about effectiveness.
When it comes to regulatory decisions about product effectiveness, FDA’s framework suggests that RWE can be used to support changes to labeling about product effectiveness, including adding or modifying an indication, such as a change in dose, dose regimen, or route of administration, adding a new population, or adding comparative effectiveness data.
So, where does that leave sponsors who want to compare the results of a single-arm clinical trial to a real-world historical control arm to demonstrate the effectiveness of a new product? Unfortunately, the FDA has set a very high bar.
Regulatory “Fitness” in Rare Disease Clinical Trials
At a joint FDA-National Institutes of Health workshop in May 2022, titled “Regulatory Fitness in Rare Disease Clinical Trials,” Katie Donohue, Director of the Division of Rare Diseases and Medical Genetics in the Center for Drug Evaluation & Research, said that the challenges facing sponsors attempting a single-arm approach to develop a first therapy for a rare disease are so daunting that development programs only “work when you are very lucky.” In particular, she pointed out that single-arm studies are vulnerable to changes in rare disease natural history.
Changes in natural history, response assessment, and standard-of-care therapy can have a dramatic effect on time-to-event endpoints such as overall survival (OS). So, for a single-arm trial, FDA recommends concrete, confirmed endpoints “like an x-ray or blood test.”
These comments highlight the strong preference FDA has for RCTs, in general, and even for rare diseases, where it is often extremely challenging to conduct an RCT with sufficient statistical power to demonstrate effectiveness.
FDA Has Set the Bar Very High
FDA’s draft guidance, titled Considerations for the Use of Real-World Data and Real-World Evidence to Support Regulatory Decision-Making for Drug and Biological Products, acknowledges the potential utility of using RWD in interventional studies, including “to serve as a comparator arm in an external control trial.” However, the guidance focuses heavily on the use of RWD/RWE in non-interventional studies, such as observational cohort studies and case control studies that evaluate the safety and effectiveness of a product in routine medical practice and are not governed by a research protocol.
Although FDA is not opposed to the idea to using RWD to construct historical control groups – also referred to as an external control arm – it is highly critical of that approach as the basis for regulatory approval of a novel drug.
Recently, Y-mAbs Therapeutics found this out the hard way. In collaboration with Memorial Sloan Kettering Cancer Center (MSKCC), Y-mAbs has developed a targeted radiolabeled antibody called 131I-omburtamab for the treatment of neuroblastoma that has metastasized to the central nervous system (CNS). This ultrarare pediatric indication affects only about 20 patients per year in the United States, and there are no approved therapies.
With traditional treatment approaches – surgery, radiotherapy (RT), and chemotherapy – most patients only survive a few months after diagnosis of CNS metastases. For about one third of patients who survive long enough to receive 2 or 3 treatment modalities, median survival is about 15 months. So, the clinical team at MSKCC, led by Dr. Nai-Kong Cheung and Dr. Kim Kramer, developed 131I-omburtamab, an anti-B7-H3 antibody, which they inject directly into the cerebrospinal fluid via an Ommaya catheter, as an adjunct to standard therapy. The goal is to eradicate residual tumor cells and increase the chance of achieving a cure.
The team at MSKCC has been studying the safety and effectiveness of 131I-omburtamab in this poor-prognosis patient population since 2004. In that timeframe, they have treated more than 100 children with CNS neuroblastoma, of whom about 40% have survived more than 8 years. Their treatment protocol demonstrated a median OS of 51 months, a milestone that clinical experts consider quite extraordinary.
Fast Forward to 2015
In 2015, Thomas Gad, whose daughter was successfully treated at MSKCC for CNS neuroblastoma, founded Y-mAbs Therapeutics to further develop 131I-omburtamab and get it approved in the US, so other children could have access to this potentially lifesaving drug.
To demonstrate the effectiveness of 131I-omburtamab, the company conducted its own single-arm multicenter trial in 50 patients, principally to confirm the results from the single-institution MSKCC trial and demonstrate objective responses to the drug. Given the rarity of this indication, an RCT was not feasible.
Y-mAbs then set out to obtain patient-level data from children with CNS neuroblastoma treated outside MSKCC and construct an external control arm for comparison with the MSKCC trial population. They succeeded in identifying only one suitable database, a neuroblastoma registry in Germany, and they were able to extract patient-level data from 120 patients who had a first recurrence of neuroblastoma in the brain. In collaboration with the FDA, Y-mAbs designed the comparative analysis using a propensity score model.
After carefully balancing the intensity of standard treatment with surgery, RT, and chemotherapy (modality group 2), the Y-mAbs biometrics team identified a cohort of 34 patients from the external control arm that they could compare to 89 patients in the MSKCC study. A comprehensive propensity score model that controlled for potential confounding factors demonstrated a 42% improvement in OS (hazard ratio = 0.58) compared with the external control arm. Sensitivity analyses showed a consistent treatment effect (hazard ratios ranged from 0.42 to 0.66) in favor of 131I-omburtamab.
Y-mAbs also went one step further, restricting the analysis to only patients in first recurrence, adjusting the index dates to control for immortal time bias, and removing patients from the external control arm treated prior to 1997. That analysis, which represents the best possible match between the populations, showed a 52% improvement in OS (Figure 1).
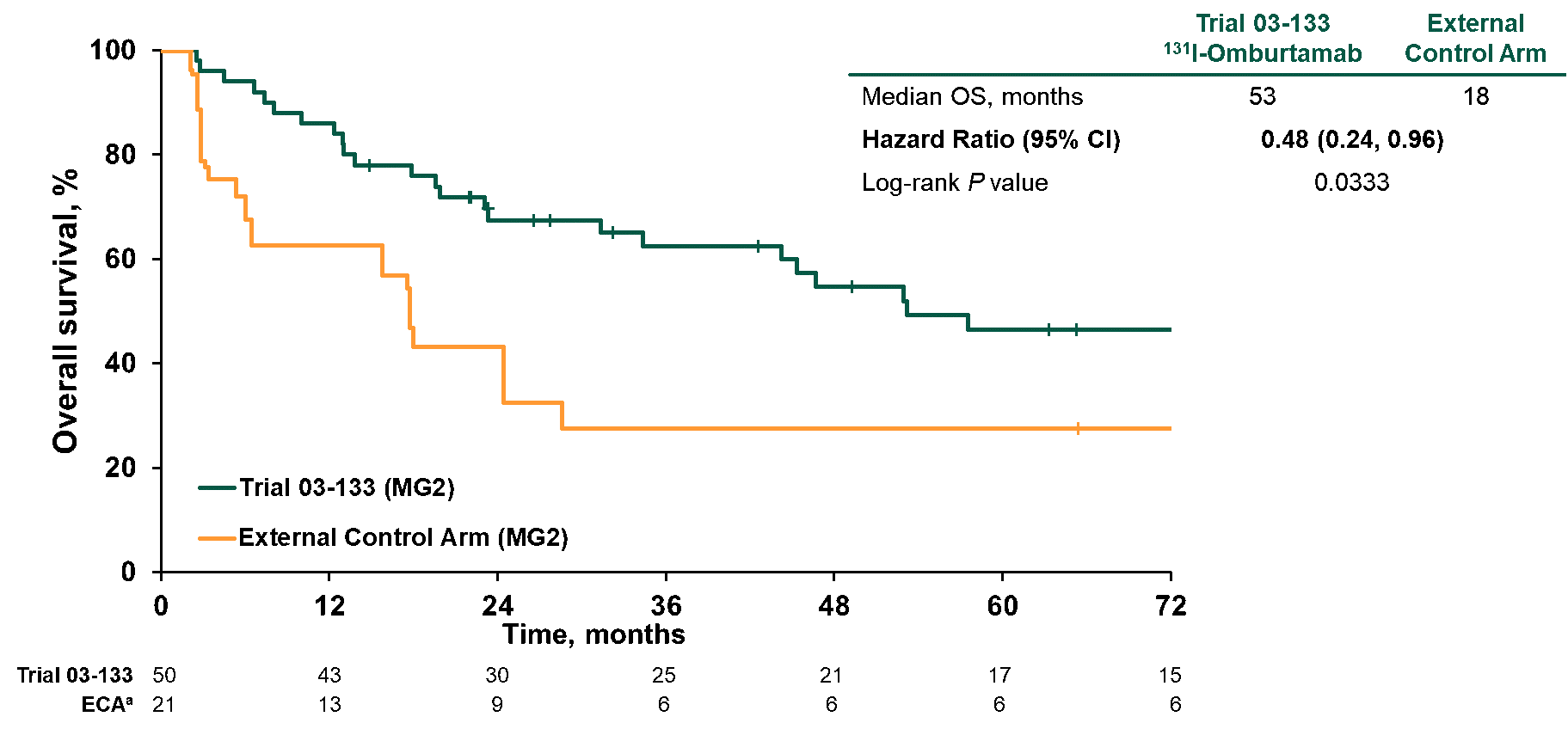
Figure 1. Overall survival in patients in modality group 2 treated at first recurrence comparing index dates A vs D and excluding NB90 from external control arm
These data, along with supportive data from the multicenter trial, were the basis for the Y-mabs Biologics License Agreement filed in March 2022. However, after careful review of the data, FDA’s Oncology Division concluded that the external control arm is “not fit for purpose.” FDA argued that limitations of the data and multiple sources of potential bias resulted in a large degree of uncertainty regarding whether the observed OS difference was due to 131I‑omburtamab or differences between the populations, or a combination of these factors. FDA also had doubts about the objective response data.
FDA Oncologic Drugs Advisory Committee (ODAC) Meeting
At the ODAC meeting on October 28, 2022, the FDA presented its case that the 2 populations were not comparable, primarily because of differences in treatment intensity and era of therapy. They pointed out that none of the patients in the external control arm received craniospinal irradiation, a form of RT perceived to be more effective than the standard focal or whole-brain RT given to the German patients. However, there are no published studies to show that it is more effective in neuroblastoma. The FDA also presented evidence that clinical outcomes for CNS neuroblastoma have improved over time. Consequently, FDA restricted its analysis to only those patients in the external control arm who were treated from 2004 to 2015, the time period corresponding to the MSKCC study.
After adjusting for all these potential confounders, including immortal time bias, the FDA analysis showed a hazard ratio of 1.0, suggesting no OS benefit.
Ultimately, the committee voted unanimously that the Applicant had not provided sufficient evidence to conclude that 131I-omburtamab improves OS in the proposed indication. The committee wanted to see more data. Unfortunately, that may not be feasible.
This case sends a strong message regarding the rigor of data that FDA expects when establishing effectiveness based on a time-to-event endpoint in a single-arm trial with comparison to an external control arm, even in a rare disease where it exercises regulatory flexibility. The consequence of this ODAC decision means that sponsors will face a high bar when attempting to demonstrate that an external control arm is “fit for purpose.”

Jeff Riegel, PhD
SVP, Scientific Communications, ProEd Regulatory
Jeff combines his scientific expertise in molecular biology and immunology with more than 25 years of global healthcare agency experience guiding medical and regulatory communication strategies for biopharma companies. Jeff helps clients prepare for FDA Advisory Committee meetings and other health authority interactions. Connect with Jeff on LinkedIn.