EMDAC meeting for Intarcia’s Diabetes Drug-Device Combo ends in rejection, but is there a silver lining?

On Thursday, September 21, 2023, the FDA held a meeting of the Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) to discuss Intarcia’s new drug application (NDA) for ITCA-650, an implantable device intended to provide continuous dosing of a GLP-1 agonist (exenatide) to treat type 2 diabetes. In a highly unusual move, Intarcia opted to request a public FDA advisory committee meeting (AdCom) in lieu of a formal evidentiary hearing following 2 Complete Response Letters (CRLs) and several formal dispute resolution requests dating back to 2017. Given this history, the prospects of swaying the panel and the FDA in favor of approval was certainly a long shot. In the end, the panel voted 19-0 that the benefits of ITCA-650 did not outweigh the risks.
It appears Intarcia was motivated not only by the prospects of a positive vote but also by the chance to present their argument for approval in a public forum. In that respect, their strategy and perseverance may have paid off. The meeting highlighted a creative use of the FDA AdCom process to communicate directly with patients and the medical community. Let’s look at how Intarcia got there.
The long path to a highly unusual EMDAC meeting: 2 CRLs, 3 rejected formal dispute resolution requests, and 1 citizen petition requesting a public AdCom
NDA #209053 for ITCA-650 (exenatide in DUROS device) was submitted on November 16, 2016. Exenatide is a GLP-1 receptor agonist, which is currently FDA-approved in formulations for subcutaneous injection twice daily or weekly. ITCA-650 is delivered by an osmotic mini-pump that is implanted under the skin and only needs to be refilled every 6 months.
The first CRL for ITCA-650 was issued by the FDA in September 2017, citing “clinical deficiencies, and device and product quality-related issues.” Shortly after the CRL, 2 ongoing trials for ITCA-650 were put on hold by the FDA due to serious safety issues, and the trials were later terminated by the company. Despite these setbacks, Intarcia resubmitted the NDA in September 2019, only to receive another CRL just 6 months later in March of 2020.
Over the rest of 2020, Intarcia filed 3 formal dispute resolution requests (FDRRs). The first 2 FDRRs were promptly denied by the Office of New Drugs (OND), reiterating unresolved safety issues, including an imbalance in severe acute kidney injury (AKI) and deaths due to major adverse cardiovascular events (MACE), as well as issues with the implanted device appropriately regulating dose. The third and final FDRR was submitted in November of 2020—this time, Intarcia requested a public FDA AdCom. The third FDRR was also rejected.
On September 2, 2021— the FDA published a notice in the Federal Register of their intention to deny the NDA for ITCA-650, and offering the opportunity for Intarcia to request a hearing. The notice outlined the agency’s reasons for the 2 prior CRLs, the 3 FDRRs, and the unresolved safety and product issues that, in their view, had never been adequately addressed. The notice detailed the conditions under which a hearing would be entertained.
It is fair to say that at this point in the process, most companies would have resigned themselves to conducting additional clinical trials or discontinuing development of the drug. Intarcia, however, did not not give up. Perhaps anticipating that their pending request for a formal hearing would ultimately be denied, Intarcia opted to request a public FDA AdCom meeting in lieu of a closed-door, formal evidentiary hearing. This request was made on February 20, 2023, via a citizen petition submitted by Intarcia, arguing that they were entitled to a public AdCom under 21 CRF part 14.
On March 24, 2023, FDA Chief Scientist, Namandje Bumpus, issued a letter granting Intarcia’s request for a public AdCom. The EMDAC meeting would be conducted under “the typical process for convening an advisory committee under 21 CFR part 14.”
So, after nearly 6 years, Intarcia would get their public meeting, but only on CDER’s terms.
EMDAC discussion centers on safety concerns, benefit-risk
The discussion centered around serious safety concerns identified by the FDA reviewers, including a higher risk of AKI, an imbalance of deaths due to MACE, and uncertainties about the drug delivery device appropriately regulating dose.
The sponsor argued that the incidence of AKI was low overall and should be understood as a class effect of GLP-1 agonists. The approved injectable formulations of exenatide (eg, Byetta and Bydureon) include warnings that the drug should not be used in individuals who have kidney disease because of increased risk of kidney injury.
The FDA also noted that the risk of death associated with MACE was unfavorable with a hazard ratio of 1.12 (95% CI: 0.83, 1.51). This was especially concerning given that other GLP-1 agonists had shown favorable results in reducing the risk of adverse cardiovascular outcomes (Figure 1).
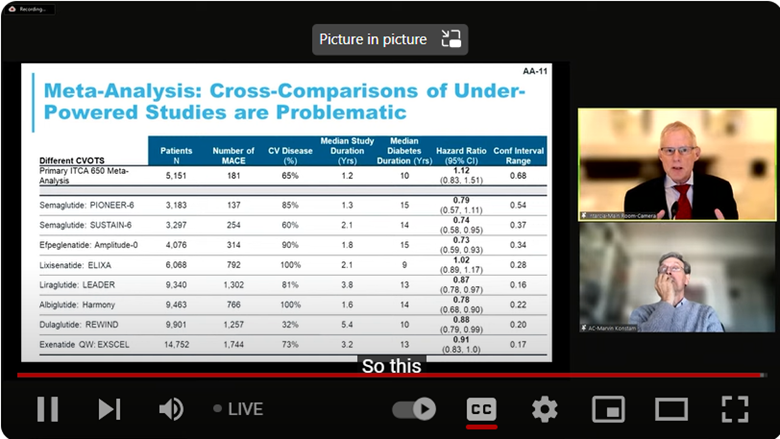
Finally, the FDA presented data showing that there was large variability in the devices delivery of the drug that could potentially lead to large amounts of drug being suddenly released into the patient. They noted that the data provided by the applicant could not rule out the possibility that adverse events were associated with inconsistencies in drug delivery. Because of these concerns, the panelists sided with the FDA, recommending that an additional safety study would be needed prior to approval to determine if the increased incidence of AKI or MACE was being caused by the implanted delivery system.
A negative vote, but a silver lining?
While a 19-0 vote against approval was surely a disappointment for the sponsor, the meeting was not entirely negative. Several panelists highlighted the superior efficacy demonstrated by the drug in controlling HbA1C levels and helping patients lose weight. During the open public hearing segment of the meeting, patient and physician testimonials were passionate and overwhelmingly positive, highlighting the benefits of ITCA-650 in improving compliance and quality of life for patients. After implantation, ITCA-650 only needs to be refilled every 6 months, eliminating the need for multiple weekly or daily injections. Several panelists were moved by these testimonials and agreed that an implantable device for diabetes treatment has potential as an important innovation.
The panelists also provided feedback regarding the types of clinical data that would be necessary to demonstrate a positive benefit/risk. For example, panelists suggested that a clinical study comparing ITCA-650 to an active control arm (such as Byetta) could help elucidate if there is a “true” increased risk of AEs with the continuous delivery mechanism compared with injectable formulations. This advice could inform future clinical trials and increase the chances of success in eventually gaining approval for ITCA-650, or similar implantable drug-devices.
Notably, Intarcia’s clinical pipeline was acquired by i2o Therapeutics in August 2023. In addition to ITCA-650, i2o is continuing development of 5 additional pre-clinical stage drug candidates for type 2 diabetes, nonalcoholic steatohepatitis, and obesity—several of which are delivered via similar implantable osmotic mini-pump technology.
While it is unlikely that the FDA will approve ITCA-650 without substantial additional clinical data, Intarcia’s decision to persevere and demand a public meeting may have a faint silver lining.
Ultimately, there are potential benefits and advantages of enduring an uncomfortable AdCom that may extend beyond simply achieving a positive vote.
 Angela W. Corona, PhD
Angela W. Corona, PhD
Senior Scientific Director, Scientific Services
Angela is responsible for helping sponsors navigate complex regulatory communications, with a focus on FDA advisory committee meetings. She develops clinical and regulatory strategy along with high-quality scientific and medical content across a wide range of therapeutic and drug development areas. Angela received her PhD in Neuroscience from The Ohio State University and completed her postdoctoral training at Case Western Reserve University.