FDA Decision for Sarepta’s Gene Therapy for DMD Sets New Regulatory Precedent

Historic Regulatory Decision for First FDA-Approved Gene Therapy for DMD
On June 22, 2023, in a-much anticipated decision, the United States Food and Drug Administration (FDA) granted accelerated approval for Elevidys (also known as SRP-9001), Sarepta’s one-time gene therapy for ambulatory children with Duchenne’s muscular dystrophy (DMD). However, the accelerated approval was limited to the subset of boys between 4 and 5 years of age who had the debilitating disease but were still able to walk. Sarepta had originally sought accelerated approval in ambulatory children aged 4 to 7 years of age, but the FDA pushed back, citing no evidence of benefit in the older patient subgroup (ages 6 to 7). Exploratory subgroup analyses are generally considered to be prone to statistical bias and/or misinterpretation; however, the FDA has used such analyses to limit indications to patients who are most likely to benefit (see our recent blog post to learn more).
While this approval is a symbol of hope for patients and families dealing with this devastating condition, the accelerated approval means that clinical benefit for SRP-9001 has not yet been established. As a condition of approval, Sarepta will be required to generate confirmatory data showing clinical benefit, including an improvement in motor function, compared with placebo. This confirmatory data will likely be forthcoming soon from the Phase 3 EMBARK trial. The FDA has promised to rapidly review these data as soon as they are available and, if necessary, adjust the indication further or recommend withdrawal of accelerated approval if clinical benefit is not confirmed.
Regardless of the ultimate outcome, this historic decision paves the way for future accelerated approvals of a rapidly growing class of gene therapies for rare diseases.
FDA sought expert advice on accelerated approval from their Cellular Tissue and Gene Therapies Advisory Committee (CTGTAC)
The FDA decision follows a May 12, 2023, meeting of the FDA’s CTGTAC where the topic of accelerated approval based on a surrogate biomarker, namely expression of micro-dystrophin (the transgene product of SRP-9001), was hotly debated. The FDA asked the committee to vote on whether the data generated from Part 2 of Study 102 were sufficient to support accelerated approval while “taking into account the existing uncertainties.”
The vote landed slightly in favor of accelerated approval, with 8 yes votes, 6 no votes, and no abstentions. However, the discussion revealed a dramatically split committee with considerable nuance associated with individual votes.
FDA commissioner, Robert Califf, has indicated that advisory committees should prioritize discussion and feedback over the strict, “thumbs-up, thumbs-down” results of the voting questions, which often receive the most media coverage. While the FDA usually makes decisions in line with the committee’s vote, it is not required to do so.
Notably, several panelists who voted “yes” revealed that they might have voted “no” on the basis of insufficient data to meet regulatory requirements but were swayed by the extremely high unmet need and the emotional testimonies of parents whose children appeared to have benefitted from SRP-9001 in videos shown during the open public hearing. Those statements, and the overwhelming unmet need likely factored heavily into the FDA’s ultimate decision to grant accelerated approval, despite major reservations on the part of FDA reviewers..
Surrogate endpoints and substantial evidence of efficacy called into question
In briefing documents released prior to the CTGTAC meeting, the FDA raised several concerns regarding Sarepta’s proposed surrogate endpoint, noting that expression of the micro-dystrophin transgene product (a purely synthetic protein not naturally expressed in skeletal muscle) does not necessarily translate into an improvement in muscle function. In other words, it is not known if expression of the transgene is “reasonably likely to result in clinical benefit” (an essential requirement for accelerated approval). This question likely cannot be answered without further placebo-controlled clinical study data.
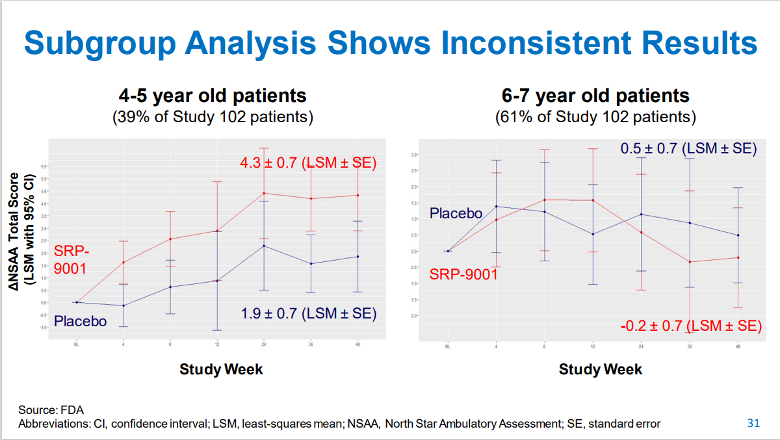
Importantly, the only randomized, double-blind, placebo-controlled clinical study (Part 1 of Study 102) failed to demonstrate a statistically significant treatment effect on muscle function versus placebo, as measured by the North Star Ambulatory Assessment (NSAA) Total Score. While an apparent effect could be discerned in the small subgroup of children aged 4 to 5 years, the study was not powered to detect a change in this group.
Slide 31 from the FDA’s CTGTAC meeting presentation (show below) illustrates these data. In the 4- to 5-year-old subgroup there was a 2.5 point difference in the change from baseline on the NSAA in patients treated with SRP-9001 versus placebo at Week 48 (P = .0172), indicating a possible clinical improvement. The 6- to 7-year-old subgroup treated with SRP-9001 had a decline in their NSAA score of -0.7 compared with placebo, indicating no apparent difference between the treatment groups. Sarepta’s scientists pointed out that an imbalance in the NSAA score at baseline may have affected the results in the 6- to 7-year-old subgroup, further confounding interpretation of the results.
The FDA concluded that
“the clinical studies conducted to date do not provide unambiguous evidence that SRP-9001 is likely beneficial for ambulatory patients with DMD. It is challenging to conclude with reasonable certainty from the data provided by the Applicant either that SRP-9001 is likely effective for younger patients, or that it is likely ineffective for older patients or those with somewhat poorer functional status. Additionally, FDA has safety concerns related to the possibility of administering an ineffective gene therapy.”
During the discussion, the CTGTAC panelists agreed that expression of the micro-dystrophin transgene speaks to the biological plausibility of SRP-9001; however, there was disagreement as to whether this expression could serve as a surrogate endpoint. Dr. Rajiv Ratan indicated, “I was not convinced, either by the nonclinical or the clinical data, that an effect on the primary functional endpoints really provided plausibility that expression of micro-dystrophin would predict clinical outcome,” and later voted “no.”
Dr. Caleb Alexander, a temporary panelist at the CTGTAC meeting and veteran of the Peripheral and Central Nervous System (PCNS) advisory committee, noted that nothing changes the fact that there was no significant effect on the primary endpoint, making it difficult to attribute any predictive power to expression of the transgene. He stated that
“…the threshold of substantial evidence has to be met whether or not a product is being approved under the standard pathway or accelerated pathways…the totality of evidence that we’ve reviewed today simply doesn’t rise to the threshold of substantial evidence that’s required for accelerated approval.”
It should be noted that the FDA has previously granted accelerated approval to 4 other therapies for DMD, three of which were developed by Sarepta – eteplirsen (Sarepta, exon 51 skipping), viltolarsen (NS pharma, exon 53), golodirsen (Sarepta, exon 53), and casimersen (Sarepta, exon 45).. These 4 therapies are all antisense oligonucleotides targeting specific mutational subtypes of DMD and use exon-skipping technology to remove a specific mutated region of the dystrophin protein to render it more functional. To date, however, none of those therapies have successfully confirmed clinical benefit, raising concerns about the feasibility of confirmatory trials in DMD.
The surrogate endpoint used in those prior approvals was expression of the “edited” dystrophin protein. The shortened versions of endogenous dystrophin produced by exon-skipping technology mimic the shortened forms of dystrophin protein found in patients who have Becker Muscular Dystrophy (a much milder form of DMD). In contrast, the expression of the SRP-9001 transgene lacks the type of empirical evidence that has been previously used to support a determination that a surrogate endpoint is reasonably likely to predict clinical benefit in DMD.
Difficulties in confirming clinical benefit following accelerated approval in DMD
Sarepta will be required to generate confirmatory data from the Phase 3 EMBARK trial to ensure that the expression of the micro-dystrophin transgene translates into significant improvement on clinical outcomes, including slowing of progression on the NSAA.
During the CTGTAC meeting, the committee emphasized the importance of the confirmatory trials to confirm clinical benefit, regardless of whether they voted yes or no, with Dr. Raymond Roos indicating that his yes vote “included the results of the [EMBARK] study that’s going to end in September 2023.”
The committee members who voted in favor of accelerated approval frequently cited the dire unmet need for treatments as their reason for voting “yes,” rather than the strength of the data package. With the added confidence that confirmatory results will be available soon, they argued that it is important not to delay treatment any longer. The dissenting committee members came to the opposite conclusion. They expressed concern that, given the uncertainty around the surrogate endpoint and the fact that a definitive answer would be available so soon, approval should only follow evidence of clinical benefit on muscle function.
Two additional considerations include the impact of accelerated approval on completing EMBARK while maintaining study integrity and what will happen if the confirmatory trial fails to generate conclusive data. Historically, withdrawal of accelerated approval after a confirmatory trial has failed has been a lengthy and complicated process resulting in potentially ineffective drugs remaining on the market for far longer than intended. We have previously written about the difficulties with confirmatory trials after accelerated approval on this blog.
Mitigating much of the concern around the confirmatory trial is the fact that the results are expected soon. The last patient, last clinical visit for EMBARK is expected to be completed by September 2023, which is very soon by accelerated approval standards. In addition, in September all patients who were on placebo are scheduled to switch over to SRP-9001. However, this does not mean that the accelerated approval will have no effect on completion of EMBARK. Patients currently in the placebo group of the EMBARK trial may demand to receive SRP-9001, rather than waiting an additional 3 to 4 months. If enough patients drop out of the study, it could reduce the ability to generate conclusive data.
In any case, the results of the confirmatory EMBARK study are highly anticipated and will determine the next steps for Elevidys. Positive results could lead to a potential conversion to full approval, along with a potential expansion of the label to include older children (6 to 7 years of age). Negative or inconclusive results, however, may result in the withdrawal of the accelerated approval.
New regulatory precedent may affect future approvals
There are currently more than 1,745 gene therapies in development, including several gene therapies for DMD (Pfizer’s gene therapy may be next in line). Indeed, gene therapy approvals seem to be gaining steam in the first half of 2023. The newest approval is Biomarin’s gene therapy for hemophilia A, Roctavian, which gained full FDA-approval on June 29. This new wave of gene therapies is promising but brings questions regarding multimillion-dollar price tags, safety of the gene delivery vectors, and longevity of transgene expression. Ultimately, many unknowns remain.
With so little precedence for gene therapy approvals, the accelerated approval of Elevidys is likely to have a major effect on future approvals for gene therapies in rare diseases. The approval establishes a regulatory precedent indicating that simply demonstrating that the transgene is expressed in the target tissue may be sufficient to use as a “reasonably likely surrogate endpoint” for accelerated approval. This indicates a willingness on the part of FDA leadership to expand the definition of what will be accepted as surrogate endpoints for gene therapies, especially when dealing with diseases of high unmet need.
 Angela W. Corona, PhD
Angela W. Corona, PhD
Senior Scientific Director, Scientific Services
Angela is responsible for helping sponsors navigate complex regulatory communications, such as FDA advisory committee meetings. She develops clinical and regulatory strategy along with high-quality scientific and medical content across a wide range of therapeutic and drug development areas. Angela received her PhD in Neuroscience from The Ohio State University and completed her postdoctoral training at Case Western Reserve University.
 Kathryn M. Madalena, PhD
Kathryn M. Madalena, PhD
Scientific Associate, Scientific Services
Kathryn provides scientific support for content development and FDA advisory committee meeting preparation across a broad range of therapeutic areas. A neuroscientist by training, with specializations in neuroendocrinology and neuroimmunology, she received her PhD in Neuroscience at The Ohio State University. Connect with Kathryn on LinkedIn.